卤键是一种被广泛应用于药物设计和材料科学等多个领域的非共价相互作用。含卤配体中的卤原子由于其电荷分布的各向异性,其顶端往往会形成一个带正电荷的亲电性区域(σ-hole),这一区域可与亲核基团相互吸引。含卤配体与蛋白质靶标之间的卤键作用已得到广泛的研究。核酸(DNA、RNA)是重要的药物靶标。由于核酸通常显电负性,这使其在理论上可以作为卤键受体,但目前尚未有含卤配体与核酸之间卤键作用的研究报道,限制了卤键针对核酸靶标的应用研究。而四分之一的上市药物含有卤素,发现含卤配体与核酸之间的卤键作用,对面向核酸的先导化合物发现与优化具有显著的理论指导意义。
在前期研究中,中科院上海药物所的研究人员发现卤键、氢键等非共价相互作用在实验解析的结构中被低估的现象(J. Chem. Inf. Model., 2017, 57, 22; J. Chem. Inf. Model., 2017, 57, 1529; J. Chem. Inf. Model., 2019, 59, 3389)。科研人员推测含卤配体与核酸之间也可能存在卤键作用,但在实验解析的结构中被低估。他们通过数据库统计分析、QM/MM优化与能量计算,发现在实验解析的结构中,核酸可以作为卤键受体与卤代配体形成卤键作用(图1),并通过自然键轨道理论和非共价相互作用等分析证实了13个核酸体系中的卤键作用。这为后续针对核酸的先导化合物发现和优化过程中,引入卤素来提高活性和选择性,奠定了理论基础。
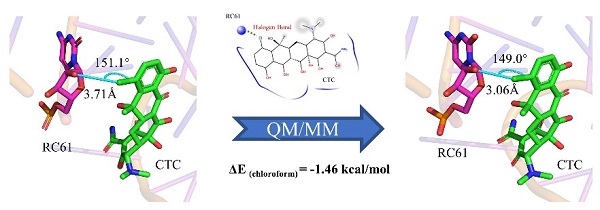
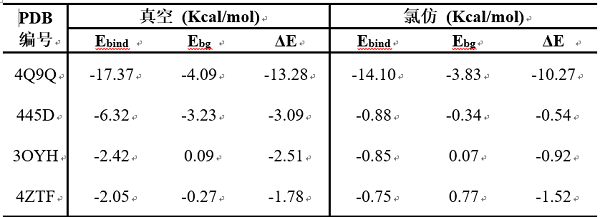
探索含卤化合物与核酸之间可能形成的卤键作用,对提高化合物-核酸之间的亲和力、改善药物的成药性、以及探索小分子配体和核酸之间的作用机制都有非常重要的作用。虽然直接通过数据库统计分析,研究人员并没有发现核酸和非共价配体之间的典型卤键作用,但他们通过对可能的核酸卤键体系进行QM/MM结构优化和作用能计算,发现13个体系中存在卤键作用(图2,表1,仅展示作为例子的4个体系)。最后,通过自然键轨道(NBO)理论和非共价相互作用(NCI)等分析,科研人员进一步证实了核酸体系中存在的卤键作用(表2,图3)。
该项研究成果近期发表于美国化学会出版的药物化学核心期刊Journal of Medicinal Chemistry。论文第一作者为上海药物所与中国科学技术大学联合培养硕士研究生穆凯洁,通讯作者为上海药物所朱维良研究员与徐志建研究员。
本工作也是该团队“分子间作用力在药物设计中的应用”系列工作的最新成果。该团队从2008年起运用数据库统计及计算化学等技术手段,对含卤药物与靶蛋白间的卤键作用开展研究,发现卤键广泛存参与含卤药物与蛋白质的结合作用(J. Med. Chem., 2009, 52, 2854; Phys. Chem. Chem. Phys, 2010, 12, 4543),可影响化合物的成药性(J. Chem. Inf. Model., 2014, 54, 69),并应用卤键作用开展先导化合物的活性和成药性优化(J. Med. Chem., 2011, 54, 5607),开展含卤老药用途的重定位研究(Repositioning)(Sci. Rep., 2016, 6, 31074)等。还研究了带负电的卤键(J. Phys. Chem. B, 2014, 118, 14223; J. Phys. Chem. B, 2016, 120, 8784; J. Phys. Chem. B, 2016, 120, 610),阐明了卤键的本质和特征(Phys. Chem. Chem. Phys., 2019, 21, 15106; J. Chem. Inf. Model., 2020, 60, 2683),并探讨了三氟甲基在药物设计中的作用(J. Chem. Inf. Model., 2020, 60, 6242)。
图1. 探索核酸体系中卤键作用的计算策略。
图2. QM/MM优化前后4个核酸体系中“卤键”几何结构的变化。其中,灰色为初始结构,绿色为优化后的结构。初始结构中的相互作用参数(距离、角度)以桔色表示,优化后的以黑色表示。
表1. 4个核酸体系中净卤键结合自由能计算
表2. 自然键轨道理论分析卤键形成的电子转移
图3. 4个核酸体系的NCI等值面,其中蓝色圆盘表示较强的吸引作用。