

中国科学院上海有机化学研究所在化学-酶催化高效合成9,10-开环甾体天然产物研究中取得进展
文章来源:上海有机化学研究所 | 发布时间:2024-02-26 | 【打印】 【关闭】
甾体作为仅次于抗生素的第二大类药物,在医药健康等领域扮演着十分重要的角色,9,10-开环甾体(9,10-secosteroids)是一类重要的亚家族,主要来源于海洋柳珊瑚,具有抗病毒、抗炎、免疫调节、抑制肿瘤细胞相关蛋白激酶等多样的生物活性。近期,中国科学院上海有机化学研究所生命过程小分子调控全国重点实验室的多个团队密切合作,报道了一种化学-酶催化的合成策略,从易得的甾体底物出发通过3~8步便可简洁、高效地合成10种9,10-开环甾体天然产物(图1)。
研究人员受到细菌甾醇降解途径的启发,策略性地提出以高效、高选择性的甾体C9α-羟基化和C9-C10键断裂为关键步骤的9,10-开环甾体合成策略。虽然多种甾体羟基化方法学被发展,但是受到空间位阻等因素影响,实现甾体C9α-羟基化依然存在很大挑战。经典的化学方法要实现甾体C9α-羟基化往往需要使用导向基、苛刻的反应条件或多步转化,合成效率很低。为了高效实现甾体的C9α-羟基化,研究人员通过筛选和比较研究,从红球菌(Rhodococcus rhodochrous)中找到了一个Rieske型非血红素单加氧酶KshA3/KshB(KSH),可以非常专一、高效地实现甾体C9α-羟基化;而当甾体底物A环为共轭二烯酮时,羟基化产物会自发发生C9-C10键断裂和A环芳构化重排反应,直接生成9,10-开环甾体产物。为了研究酶催化反应机制,研究人员进行了酶的反应动力学、结构预测、分子对接计算和点突变等实验,提出了酶通过关键氨基酸Y226、N251残基及水分子介导的氢键网络来识别和定位甾体底物,从而实现酶高效性和专一性地催化甾体的C9α-羟基化。
基于发展的酶催化甾体C9α羟基化-碎裂串联反应(9α-hydroxylation and fragmentation cascade),研究人员进行了底物拓展研究,发现该酶不仅可以兼容各种不同C17位取代的共轭双烯酮底物,还能兼容共轭单烯酮底物从而获得非开环重排的C9α-羟基化产物,产率最高可达97%,并且能进行克级规模放大。基于设计的合成策略,研究人员利用化学-酶催化方法,通过3~8步便可实现10种9,10-开环甾体天然产物的高效、快速合成。进一步的抗菌活性测试及构效关系研究表明,该类化合物对耐万古霉素粪肠球菌(Enterococcus faecalis)具有抑制活性。该研究显示了化学-酶法合成策略的潜力和重要性,促进了9,10-开环甾体生物合成的机制理解,为高效合成该类产物提供了新的方法,并为进一步的生物学功能研究提供了物质基础。
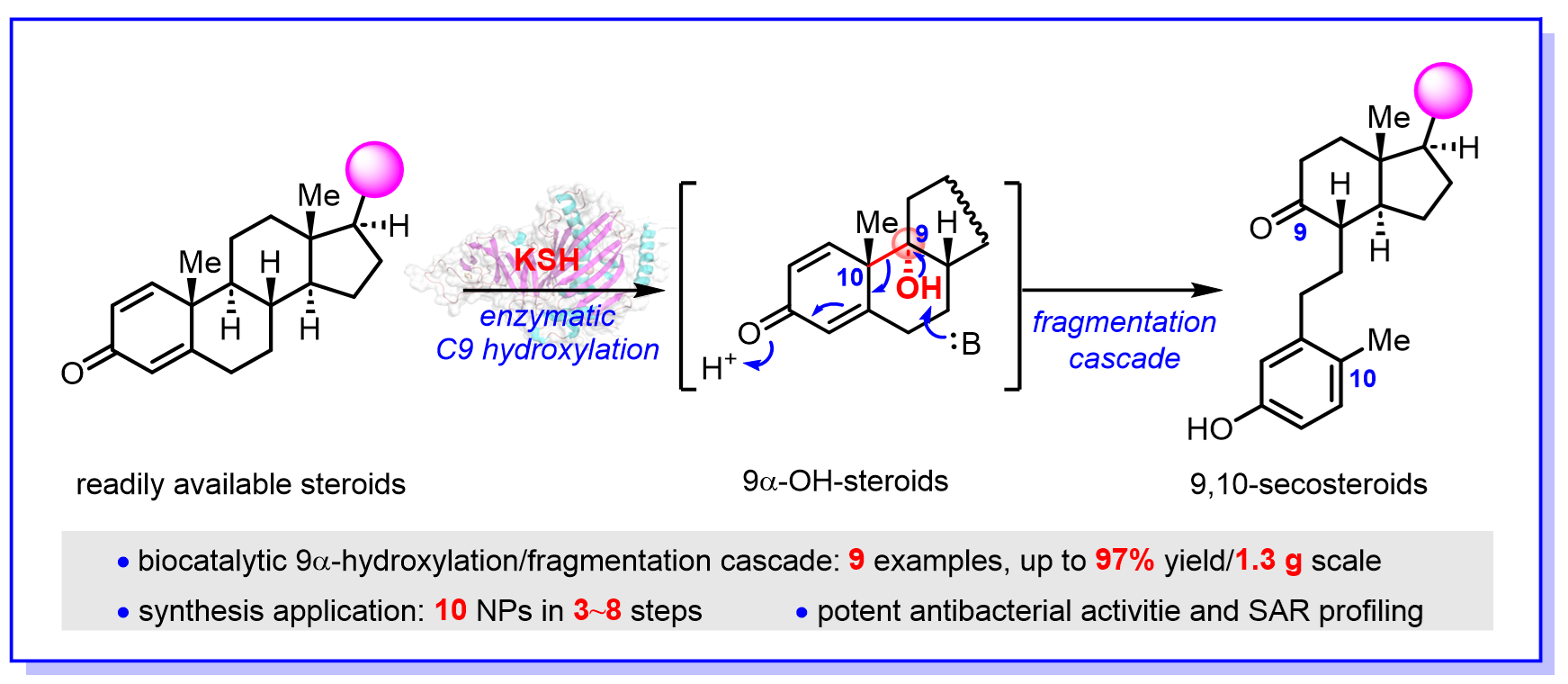
图1 生物催化甾体的C9-羟基化和碎裂串联反应实现9,10-开环甾体高效合成
该研究成果近期在“Angewandte Chemie International Edition”上发表(DOI:10.1002/anie.202319624)。有机所博士研究生宋瀚鑫和张泽良为论文的共同第一作者,曹春阳研究员、汤志军副研究员、桂敬汉研究员和刘文研究员为通讯作者。该工作得到了科技部、国家自然科学基金委和新基石科学基金会的大力资助。
